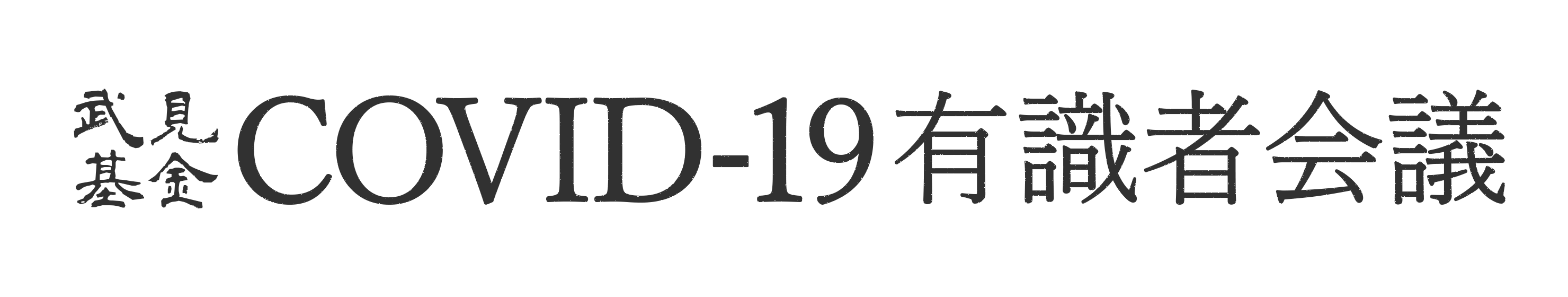
- COVID-19治療薬の開発は急務であり、基礎研究に基づいたドラッグリポジショニングを、ワクチン開発と並行して進めるべきである。
- 筆者らは、2016年MERS-CoVのSタンパク質による膜融合を、DSPレポーターで定量的モニターするスクリーニング系を確立した。さらにFDA承認既存薬ライブラリーから、膜融合を強力に阻害する薬剤としてナファモスタットを同定した。
- ナファモスタットは、急性膵炎や播種性血管内凝固症候群に長年使われてきた既存薬である。安全性に関する臨床データが蓄積し、薬物動態も解析されている。
- コロナウイルス(CoV)は、ウイルス遺伝子が脂質二重膜とSタンパク質からなる外膜で囲まれており、感染にはSタンパク質によって誘導される外膜と細胞の膜(細胞膜やエンドゾーム膜)との融合が必須である。ナファモスタットは気道細胞表面にあるタンパク質分解酵素TMPRSS2を抑制することにより、COVID-19の原因ウイルスSARS-CoV-2のSタンパク質による膜融合阻害した。ナファモスタットは、気道上皮細胞由来のCalu-3細胞へのSARS-CoV-2感染を、EC50=10 nMで抑制した。
- ナファモスタットは、持続点滴静注により平均140 nMの血中濃度を維持できることから、COVID-19治療の抗ウイルス剤として期待できる。
- ナファモスタットは、血栓形成や炎症性サイトカインの産生を抑制する。このためCOVID-19の重症化阻止も期待されるが、有効性は臨床試験により検証する必要がある。
はじめに
新型コロナウイルス(severe acute respiratory virus coronavirus 2 (SARS-CoV-2))が原因となる感染症(coronavirus disease 2019 (COVID-19))は、昨年暮れに武漢で、世界最初の患者が確認された。世界保健機構(WHO)も3月11日にパンデミックを宣言した。ほぼ半年が過ぎ、世界の感染者約870万人、死者46万人以上、国内でも感染者約1万8000人、死者950人以上(2020年6月21日現在)に達した[1, 2]。感染者の中には一定の割合で無症状(不顕性)の方がおり、症状がある人の80%程度が軽症であるものの、重症化したり、さらに高齢者や基礎疾患がある人の場合を含めて、死に至ることがある。しかしながら現時点で、国内で承認されている治療薬はレミデシビルのみであり、依然としてCOVID-19に対して効果が確認された治療薬の開発は急務である。しかも既に全世界的に感染拡大している現状を鑑みると、安全性が確認された既存薬から治療薬を探すいわゆるドラッグリポジショニングがワクチン開発と並行して進められるべきである。このような緊迫した背景の中、ナファモスタットがアカデミアの基礎研究からCOVID-19治療薬のシーズとして提案され、臨床研究に至った経緯について述べたい。
医科学研究所 北京日中連携研究室 -感染症の基礎研究に最高の環境-
筆者が所属する東京大学医科学研究所(医科研)は、2005年からアジア感染症拠点を設置している。北京においては中国科学院生物物理研究所および微生物研究所の各々に日中連携研究室を設け、そこに医科研の教員が常駐することで、中国研究者との綿密な連携のもと感染症の研究を進めてきた[3]。本拠点は、初めの2期10年間は文科省(第1期2005~2009年度「新興・再興感染症研究拠点形成プログラム」、第2期2010~2014年度「感染症研究国際ネットワーク推進プログラム」)、次の5年間はAMED(第3期2015~2019年度「感染症研究国際展開戦略プログラム」)の支援により運営されてきた。筆者は、第2期の途中2011年から、日中連携研究室のメンターとして年に3〜4回北京を訪問して常駐教員に研究や運営状況について助言をしてきた。筆者の専門は、細胞内シグナル伝達で、特に乳がんの悪性化における転写因子NF-κBの役割について研究していた。もともと大学院の時にセンダイウイルスによる膜融合機構の研究に携わり、卒業後も癌研究会癌研究所で、成人T細胞白血病の原因ウイルスであるヒトT細胞白血病ウイルス1型(HTLV-1)による白血病発症機構について研究に従事した。
このため感染症研究には人並み以上のアフィニティーを持っていた。このメンターは研究所の業務であるものの、自身の興味も手伝って感染症研究に積極的に関与していた。ただ、この9年後にSARS-CoV-2の膜融合の研究をするとは夢にも思っていなかったが、2002年〜2003年にかけて中国がSARS-CoVの感染拡大の経験しており、両研究所においてコロナウイルスに関連した研究が進行していたことや、連携した両研究所が感染症学と構造生物学の世界トップレベルの研究所であり、一流の基礎研究が進められていたことが、その後、ナファモスタットの研究に至った大きな要因の一つである。当時、日中連携研究室の主なテーマはHuman Immunodeficiency Virus 1 (HIV-1) の膜融合を介した細胞侵入と潜伏感染の機構解明であった。HIV-1のようにウイルス遺伝子が脂質二重膜と膜タンパク質からなる外膜(エンベロープ)で囲まれているウイルス(「外膜ウイルス」または「エンベロープウイルス」と呼ぶ)は、外膜と細胞の膜(細胞膜やエンドゾーム膜)とが融合することで、ウイルス遺伝子が細胞質に移動し感染が成立する。常駐教員の一人である松田善衛特任教授は、HIV-1の侵入過程であるウイルス外膜と細胞膜との融合を分子レベルと解明しようとしていた。松田先生のアイデアがこの後の日中連携研究室の研究を支えていくことになる。
膜融合を定量するDSPレポーターの開発
Dual Split Protein (DSP) は、発光タンパク質Renilla luciferase(RL)のN末端側断片と蛍光タンパク質GFPのN末端側断片を結合させたキメラタンパク質(DSP1-7)と、GFPのC末端側断片とRLのC末端側断片を結合させたキメラタンパク質(DSP8-11)を用いる【図表1】。DSP1-7とDSP8-11は、ともに単独では蛍光を発しないし発光もしないが、両者が出会うとGFPの分割部分同士がすぐに再集合する。同時にRLの部分も再構成して両キメラタンパク質は安定な複合体を形成し、蛍光を発し発光するようになる[4, 5]。
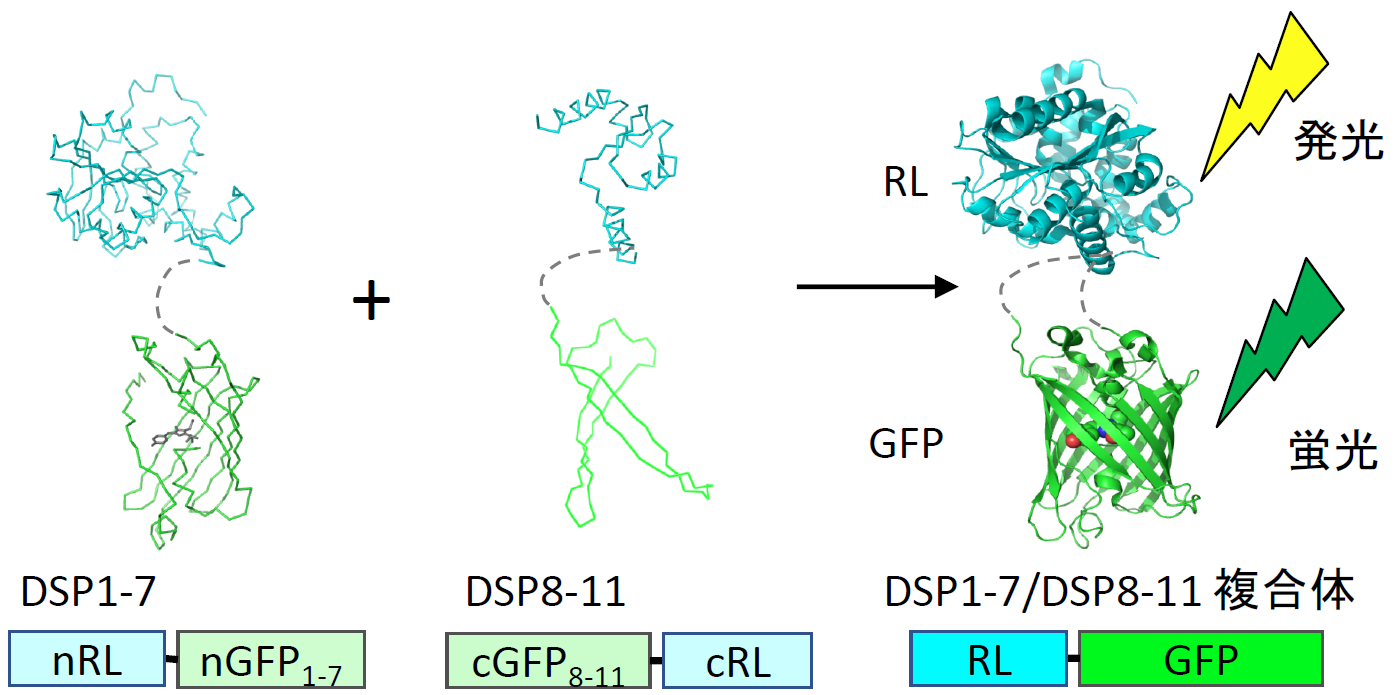
例えばHIV-1の場合、受容体結合と膜融合能を持つ外膜タンパク質であるgp120 とgp41およびDSP1-7を293FT細胞に発現させてエフェクター細胞を作成する。一方で別の293FT細胞に受容体であるCD4と副受容体CCR5およびDSP8-11を発現させて、ターゲット細胞を作成する。このエフェクター細胞とターゲット細胞を混合すると、細胞融合が起こるとともに蛍光が生じ発光する【図表2】。
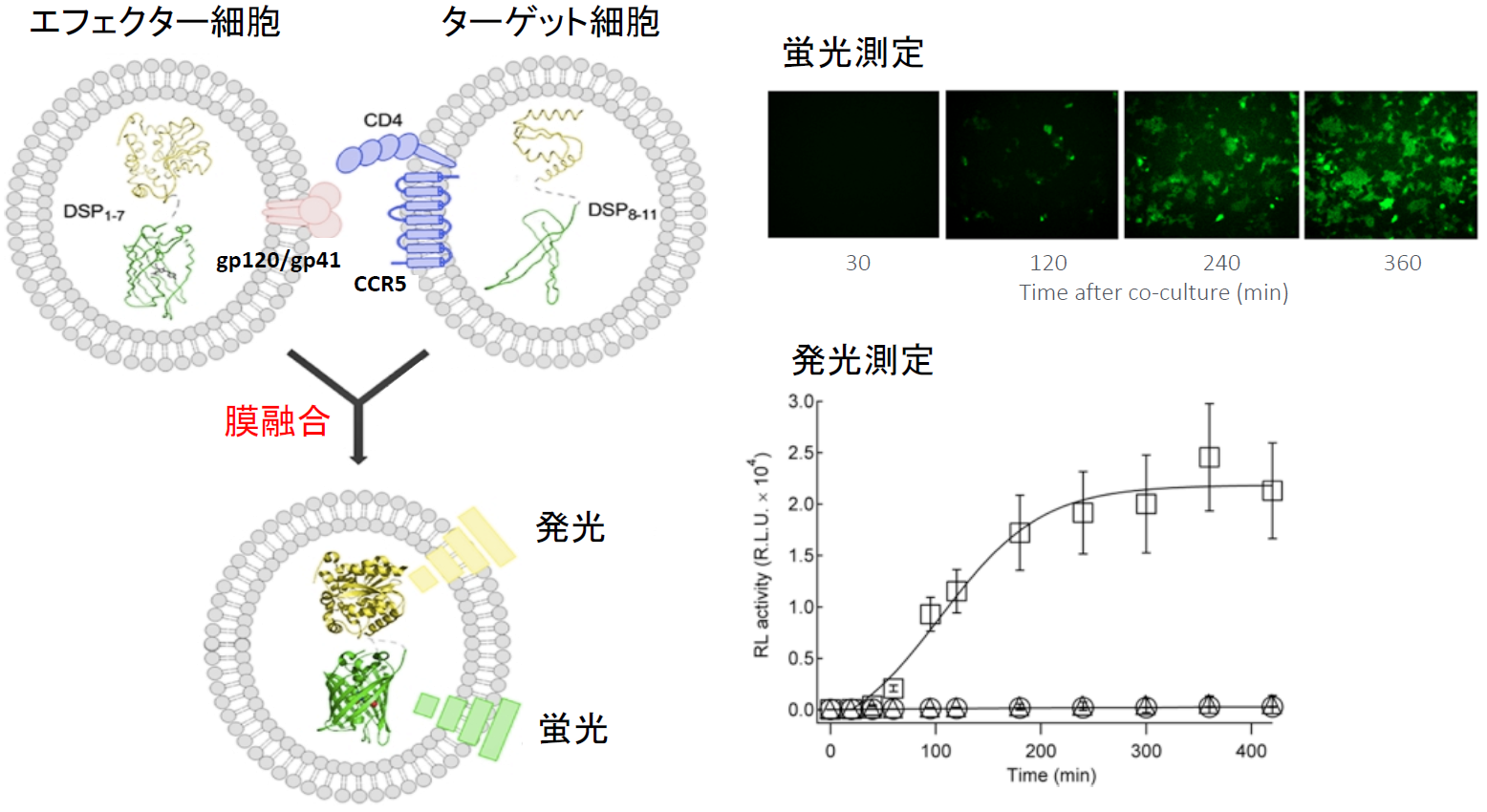
筆者らは通常、定量性と感度の良さから発光により融合を定量化している。この融合が非特異的なものでなく、ウイルス外膜タンパク質と受容体との相互作用に依存する特異的なものであることは、外膜タンパク質とCCR5との結合を阻害する化合物Maravirocを加えると、融合を示す発光量が抑制されることから判断できる【図表3】。
さらに、Maravirocは実際に抗HIV-1剤として承認されている。ということは、松田先生が確立した細胞-細胞融合をDSPリポーターで定量する実験系は、融合の分子メカニズム研究ばかりでなく、融合阻害を作用点とする抗ウイルス剤の同定にも応用できることを示している。実際、筆者らはこのDSPリポーターを用いて、今回のSARS-CoV-2だけでなくジカウイルスやデングウイルスに応用して、新規の感染阻害化合物を既存薬から見出している(投稿中)。基本的に膜融合が侵入経路であるウイルスには、この融合系が利用できると考えている。
| 図表1 |
| Dual Split protein (DSP) レポーターによる膜融合定量の原理 |
| DSP1-7は、Renilla luciferase(RL)のN末端側断片とGFPのN末端側断片を結合させたキメラタンパク質で RL1-155-Ser-Gly-Gly-Gly-Gly-GFP1-156の構造を持つ。DSP8-11はGFPのC末端側断片とRLのC末端側断片を結合させたキメラタンパク質でMet-GFP157-231-Ser-Gly-Gly-Gly-Gly-RL156-311の構造を持つ。DSP1-7およびDSP8-11は両方とも単独では蛍光を発せず発光もしない。両者は出会うとGFP断片同士が複合体を形成するので、luciferaseもGFPも再構成して蛍光を発し発光するようになる。原理的には多くの外膜ウイルスや、ウイルス以外の原因で起こる膜融合にも応用可能と考えている。 |
 |
| 図表2 |
| HIV-1外膜タンパク質を発現した293FT細胞と受容体CD4と副受容体CCR5を発現した293FT細胞の融合 |
| HIV-1の外膜タンパク質であるgp120 とgp41およびDSP1-7を293FT細胞に発現させてエフェクター細胞を作成し、一方で受容体であるCD4と副受容体CCR5およびDSP8-11を発現させたターゲット細胞を作成する。両細胞を37℃で混合すると細胞融合が30分以内に起こり始め、4時間ほどでほぼプラトーになる。蛍光でも発光でも測定できるが感度と定量性から発光により定量している。 |
 |
| 図表3 |
| HIV-1外膜タンパク質による細胞融合の特異性 |
| HIV-1のJRFL株は副受容体としてCCR5を使うが、HXB2株はCXCR4を使う。MaravirocはCCR5の阻害剤でJRFL株の外膜タンパク質とCCR5との結合は阻害するが、HXB2株の外膜タンパク質とCXCR4との結合は阻害しないことが報告されている。筆者らの膜融合実験では、その特異性が明確に反映されており、MaravirocはJRFLによる細胞融合は阻害するが、HXB2による細胞融合には全く影響しない。 |
 |
MERS-CoVによる膜融合阻害薬のハイスループットスクリーニング
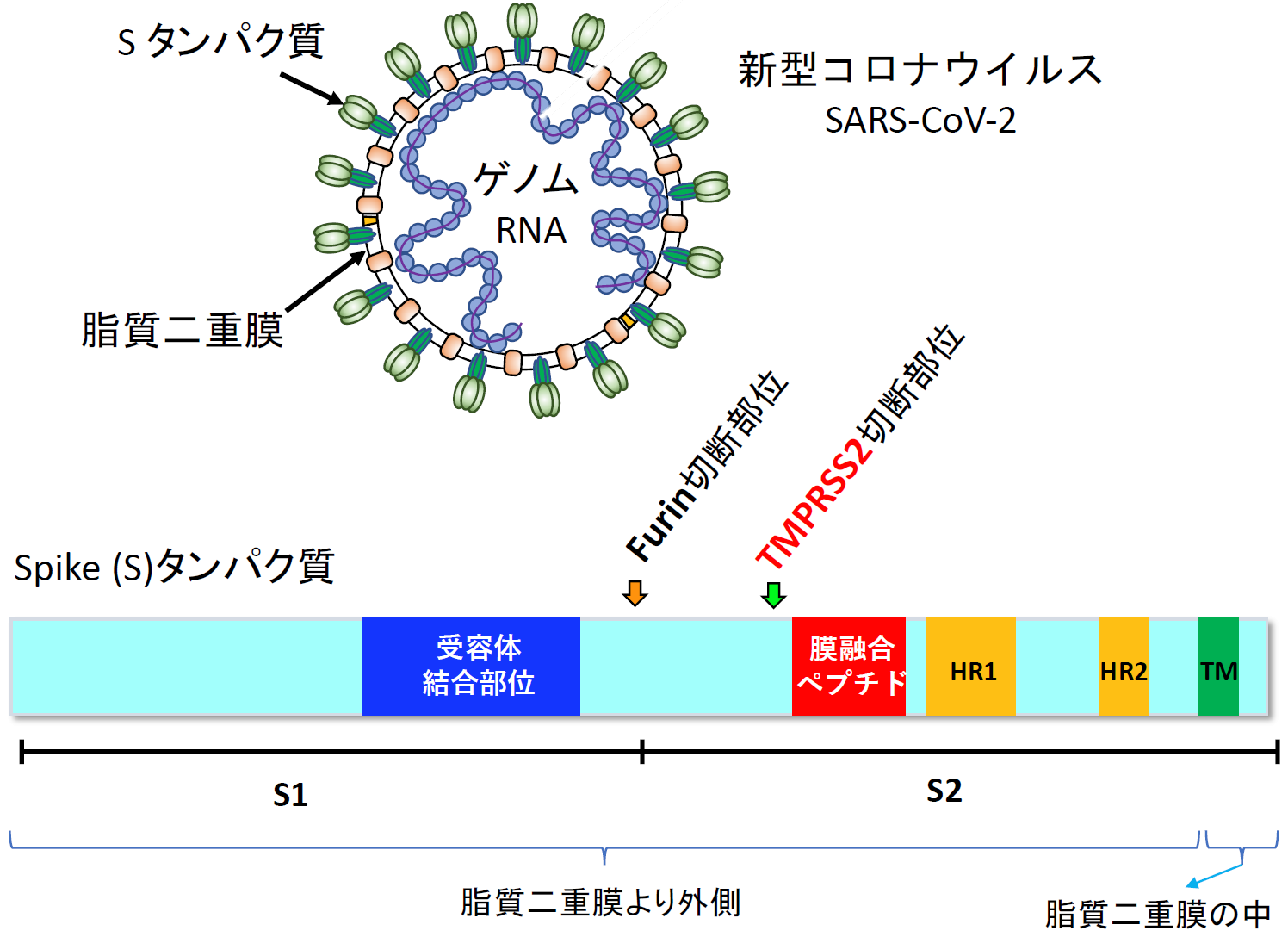
2016年ごろ、筆者らはHIV-1と同様に外膜ウイルスであるMiddle East respiratory syndrome coronavirus (MERS-CoV) の感染阻害剤の探索を始めた。なぜMERS-CoVなのか?今日のCOVID-19の出現を明確には予想はしていなかったものの、2003年のSARS-CoV, 2012年のMERS-CoVの流行の後、2015年の韓国でのMERS-CoVの再流行等の流れが、何となく新たな病原性を持ったコロナウイルスの将来的な出現を想像させていたのは事実である。もう一つの理由は、前述のように中国科学院の日中連携研究室の周りに、MERS-CoVの研究をしている研究者がいたからである。ちょうどこの頃、山本瑞生助教(現特任講師)がアジア感染症拠点に参加し、松田先生が確立した融合系を発展させナファモスタットの発見に繋がっていく。中東呼吸器症候群MERSは、肺炎を主症状とするが、無症状の例から急性呼吸窮迫症候群acute respiratory distress syndrome (ARDS) の重症例まである。2019年9月現在、感染者は2468例、死亡者は851例、死亡率34.4%前後である[6]。コロナウイルスの場合、受容体結合や膜融合に関与する外膜タンパク質はSpike (S) タンパク質と呼ばれる【図表4】[7]。
Sタンパク質は、ウイルスが感染細胞から出てくる前に細胞が持つFurinというタンパク質分解酵素により中央付近で切断され、S1とS2に分かれる(コロナウイルスの種類によっては、Furin以外の酵素の可能性や切断の頻度等が未確認である)[8]。S1タンパク質は受容体結合部位(Receptor Binding Domain (RBD))を介して気道細胞表面にあるCD26(SARS-CoV, SARS-CoV-2の場合は angiotensin-converting enzyme 2 (ACE2))という膜タンパク質を受容体として結合する【図表4、5】[9]。その後、S2がTMPRSS (Transmembrane serine protease 2) と呼ばれる気道細胞表面にあるタンパク質分解酵素により1箇所切断されてS2’となる[10]。S2のTMPRSS2切断部位の近傍には膜融合ペプチド(fusion peptide)と呼ばれる気道細胞膜に接触して膜融合を誘導する部位があり【図表4】、S2の状態では隠れていた膜融合ペプチドがTMPRSS2でS2’になることで外側に突出して膜融合を誘導すると考えられている【図表5】。そのためTMPRSS2 による切断はプライミング(priming)と呼ばれている。
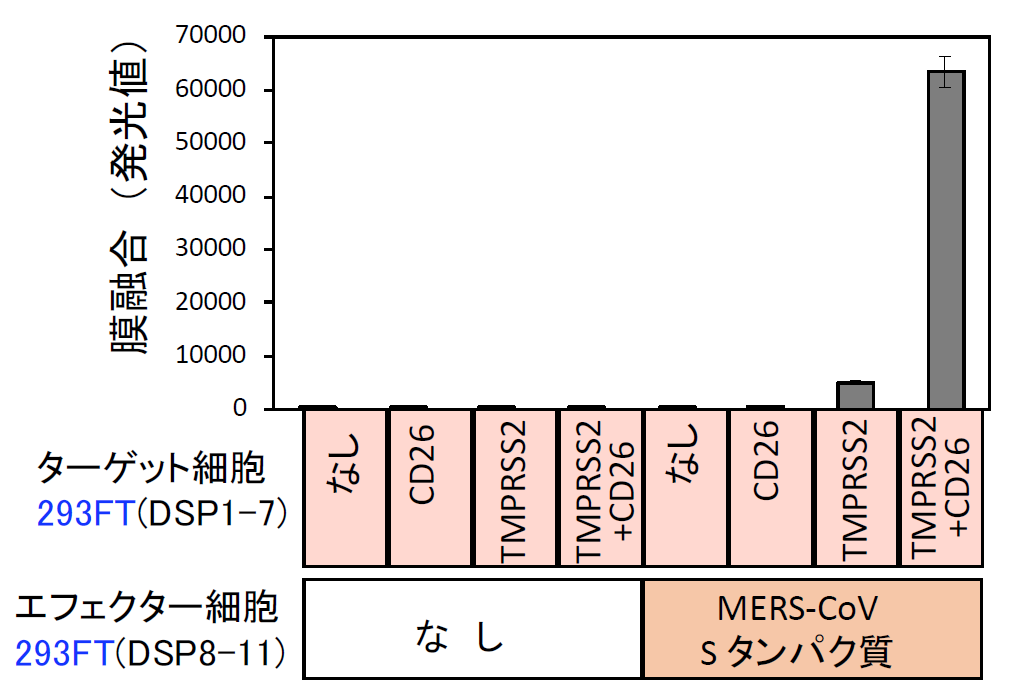
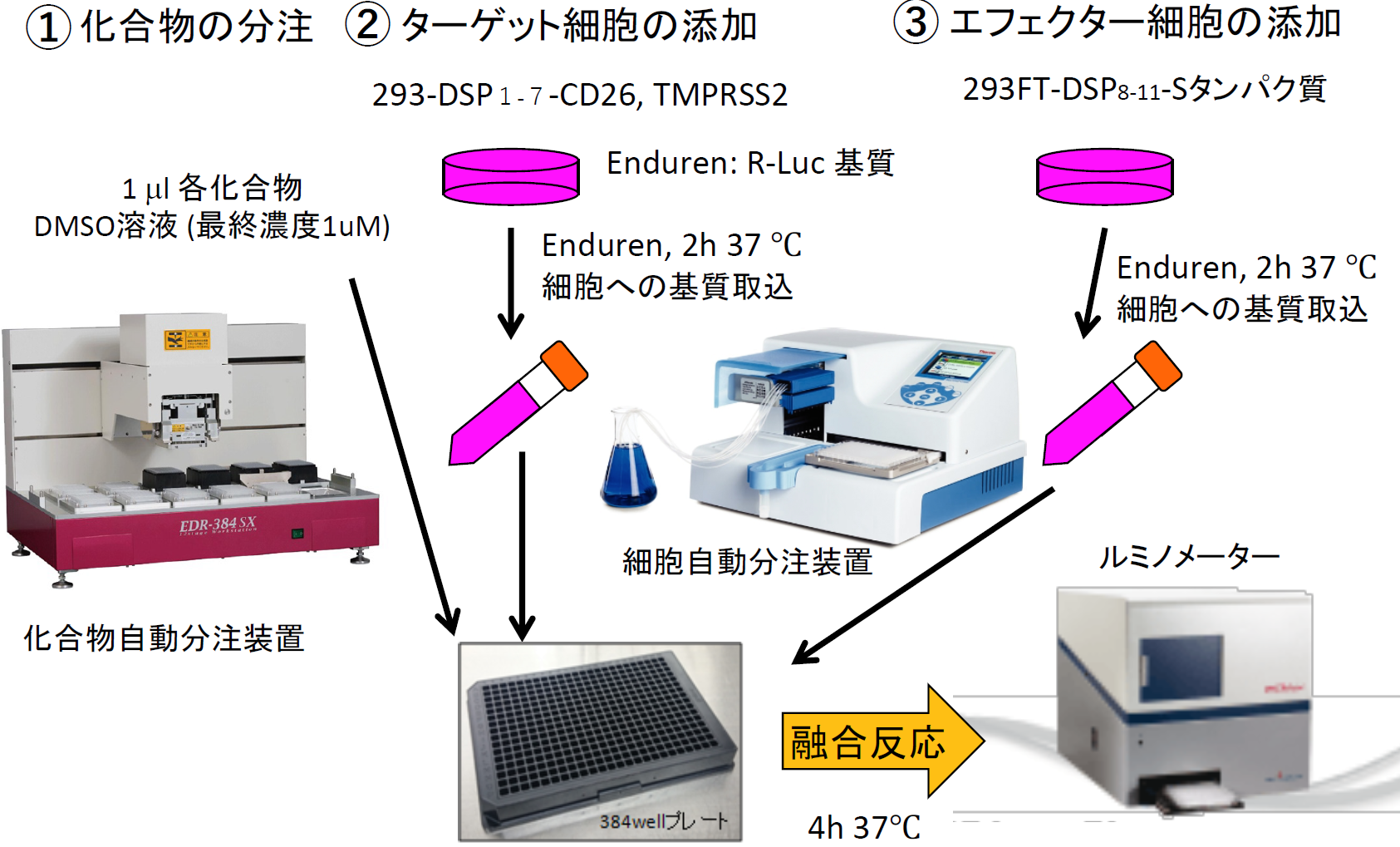
そこで、MERS-CoV Sタンパク質及びDSP8-11を293FT細胞に発現させたエフェクター細胞およびCD26とTMPRSS2及びDSP1-7を293FTに発現したターゲット細胞を作成した【図表6】。両細胞を混ぜると効率よく融合が起こり、この融合がSタンパク質、受容体CD26, TMPRSS2に完全に依存しており、本融合系の特異性を示している【図表7】。この系を利用して多数の既存薬や化合物群(ライブラリー)の融合阻害能を調べるには、数に対応するためのスケールダウンと、確実さを確保したまま時間を短縮するための機械化により、ハイスループット化をする必要がある。そのために、これまで24ウェルプレートで実験していたものを384ウェルプレートに変更し、化合物や細胞の分注を機械でできるように条件を検討し、ハイスループットスクリーニング(HTS)系の確立に成功した【図表8】。
| 図表4 |
| SARS-CoV-2 Sタンパク質の構造とTMPRSS2によるプライミング |
| コロナウイルス全般の説明をするために、SARS-CoV-2を例として示した。Sタンパク質のS1部分で受容体に結合し、その後TMPRSS2でS2に切れ目が入り(primingと呼ばれている)、S2中の膜融合ペプチドの部分が感染細胞の細胞膜に接触して、膜融合が起こると考えられている。 |
 |
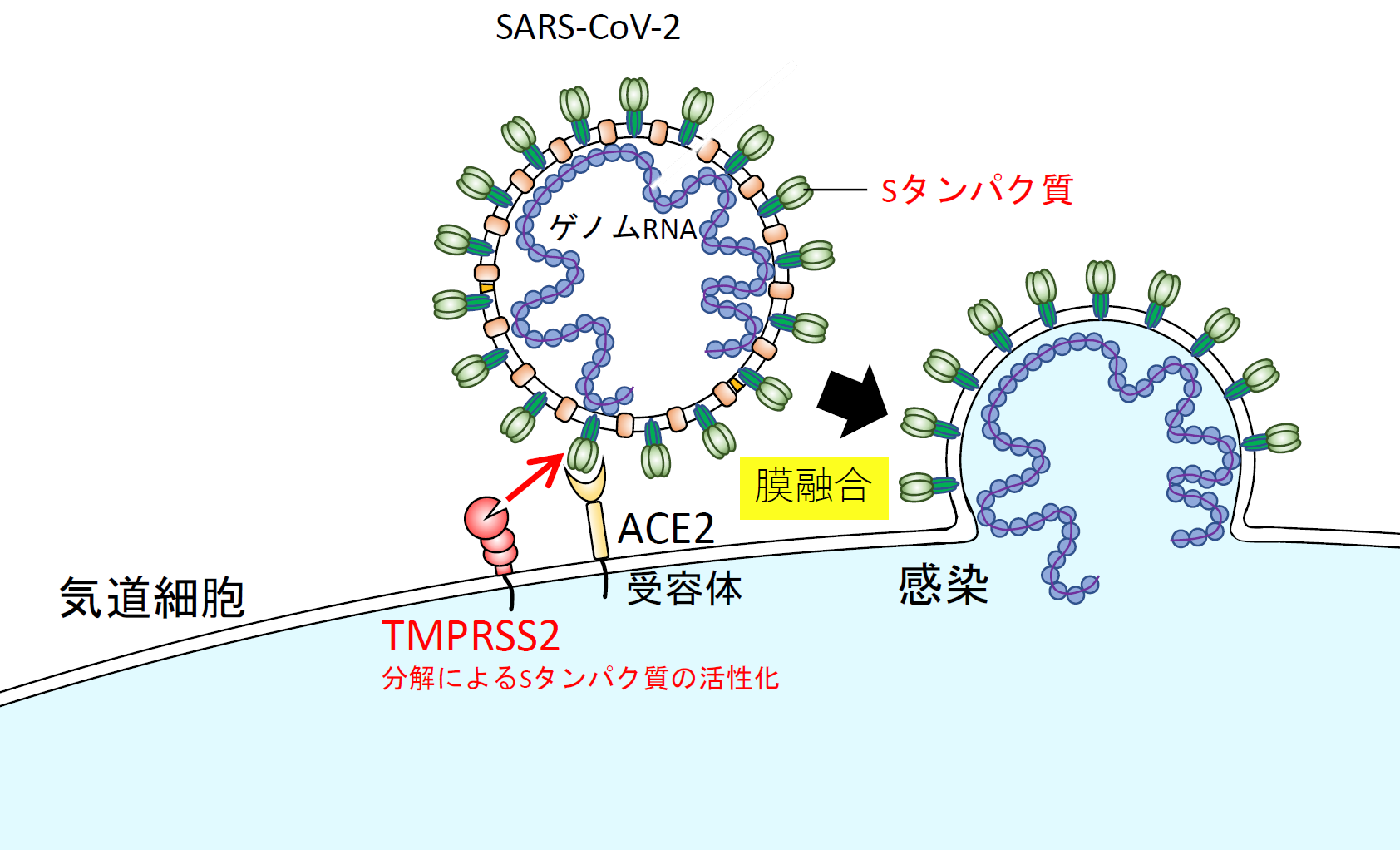
| 図表5 |
| SARS-CoV-2の膜融合を介した気道細胞への感染 |
| 【図表4】の説明で膜融合が起こるとウイルスのゲノムRNAが細胞質に移行して、ウイルスRNAの増幅が起こる。本文中で説明しているが、コロナウイルスにはTMPRSS2非依存的な感染経路がある。ウイルスが受容体結合後、エンドサイトーシスされ、エンドゾームでカテプシンによりSタンパク質がプライミングされ、ウイルス外膜とエンドゾーム膜の融合が起こり、ゲノムRNAが細胞質に移行する経路である。このことはナファモスタット の抗ウイルス作用と深く関係しており本文で解説している。 |
 |
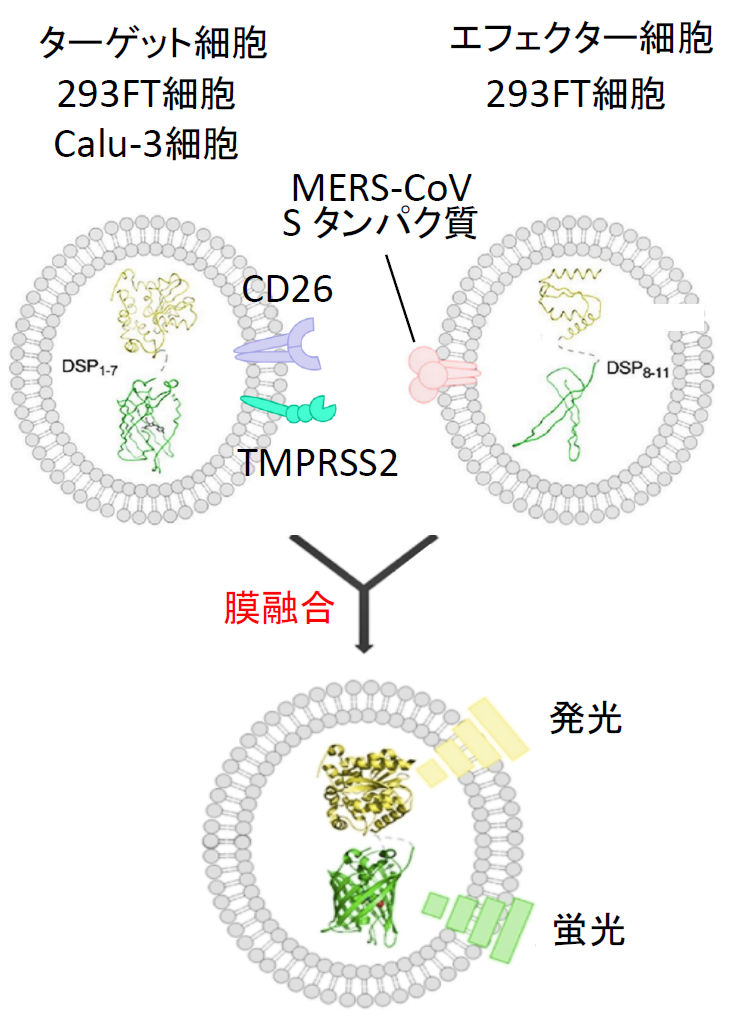
| 図表6 |
| MERS-CoV Sタンパク質による細胞融合系 |
| MERS-CoV SタンパクおよびDSP8-11を293FT細胞に発現させてエフェクター細胞を作成し、一方で受容体であるCD26とSタンパク質を活性化するTMPRSS2、およびDSP1-7を発現させたターゲット細胞を作成する。両細胞を37℃で混合すると細胞融合が30分以内に起こり始める。 |
 |
| 図表7 |
| MERS-CoV Sタンパク質による細胞融合の特異性 |
| MERS-CoV Sタンパク質による細胞融合系は、Sタンパク質、受容体CD26、TMPRSS2の全てに依存する特異性を有する。 |
 |
| 図表8 |
| MERS-CoV Sタンパク質による細胞融合の阻害剤探索のためのハイスループットスクリーニング(HTS)系 |
| HTSは、系の安定性が生命線である。スケールダウン、機械化によっても安定性を失わないために、条件検討は慎重に行った。完全に自動化しているわけではないので、人間の手技にも影響される。 |
 |
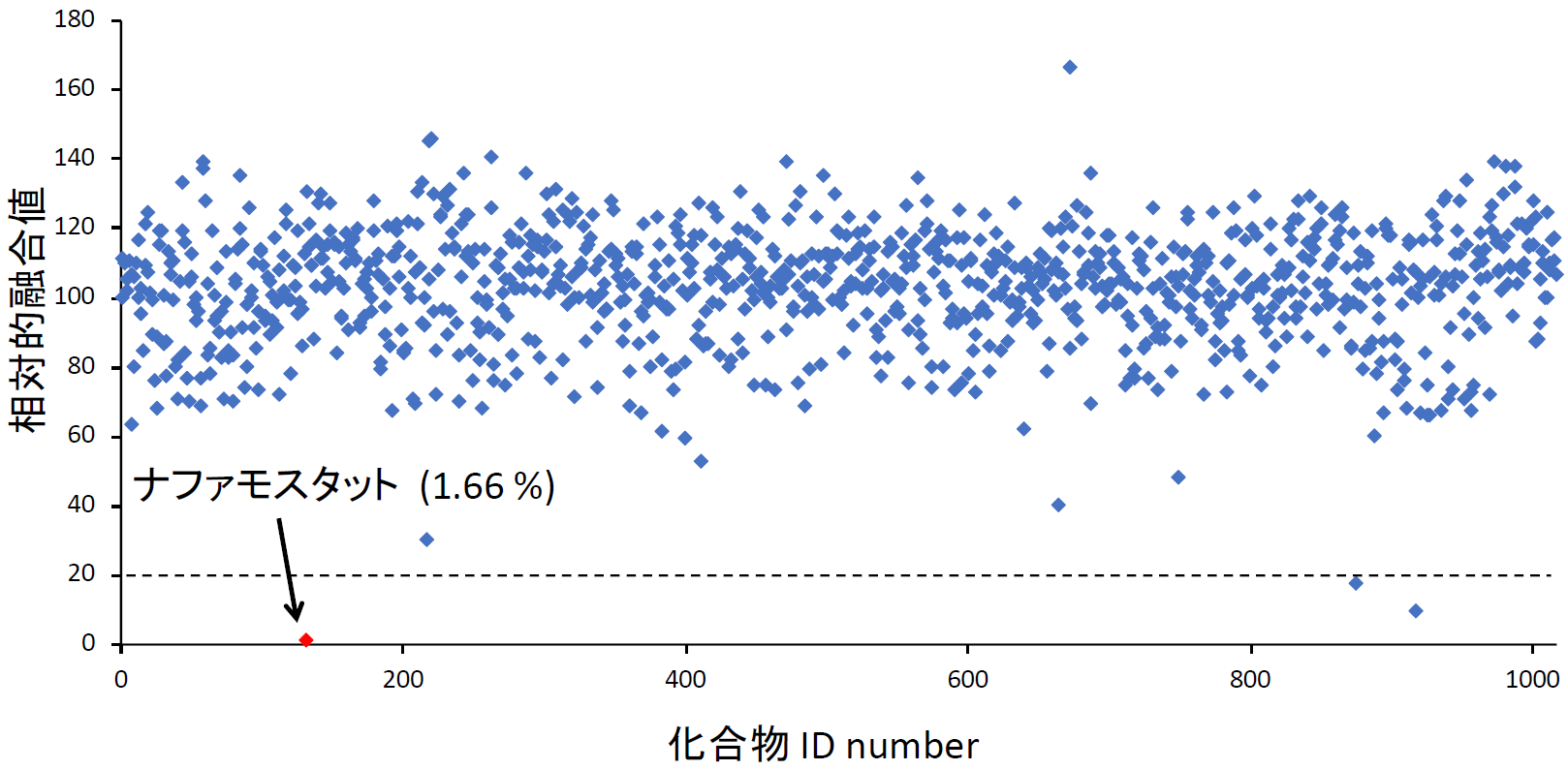
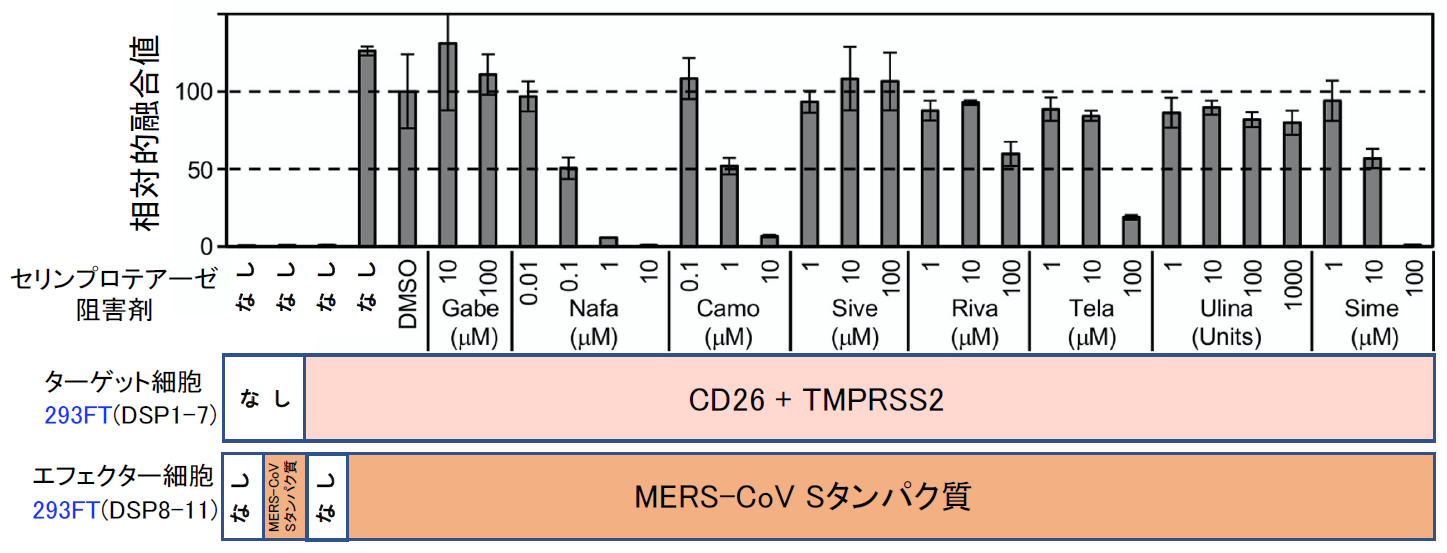
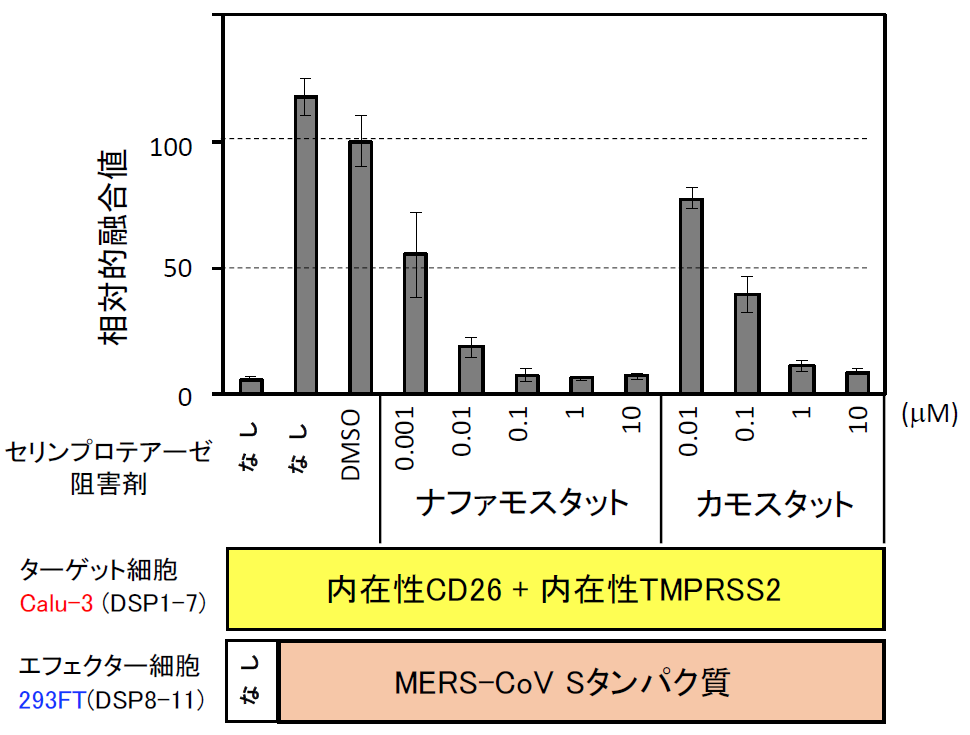
まず、Drug repositioningを念頭にこの条件で米国Food and Drug Administration (FDA) で承認された1017化合物からなるライブラリーをスクリーニングした。安全性が確保され認可されている化合物であれば、すぐに臨床試験を開始できるからである。スクリーニングの結果、全ての化合物の濃度を1 μMに揃えている条件でトップヒットが、ナファモスタットであった(【図表9】ナファモスタットの値は1.66%。化合物はDMSOに溶けている。同量のDMSOのみの場合の融合を100として%表示している。従って全く阻害能がない場合には値が100%付近になる。値が低いほど阻害能が高い)。ナファモスタット がセリンプロテアーゼ阻害剤であった[11]ので、国内で薬として認可され臨床に用いられている主なセリンプロテアーゼ阻害剤(ガベキセート、シベレスタット、リバーロキサバン、テラプレビル、ウリサスタチン、シメプレビル)の効果をみたところ、ナファモスタットが最も効果的で、次にカモスタットであり、残りの阻害剤は効果がある場合でも100 μM以上を必要とする【図表10】ことから、以降の解析はナファモスタットとカモスタットに絞って進めた。ここまでは、ターゲット細胞として、ヒト胎児腎臓細胞由来の293FT細胞に遺伝子導入により異所的にCD26と、TMPRSS2を過剰発現したものを使っていたが、これらの発現がより生理的に近いヒト気道上皮細胞由来のCalu-3細胞(内在性にCD26とTMPRSS2を発現している)を用いて、ナファモスタットとカモスタットの阻害濃度を比較した。その結果、ナファモスタットは1nMで50%程度阻害し、10 nMで90%程度阻害した。一方カモスタットでは、10 nMでは20%、100 nMで60%程度の阻害であり、ナファモスタットの方がカモスタットより10倍以上阻害能が高いことが明らかとなった【図表11】。
次に、実際の生のMERSコロナウイルスの感染をin vitroで阻害する能力を、ナファモスタットとカモスタットで比較した【図表12】。この実験は感染研の松山州徳先生、竹田誠先生との共同研究である。その結果、ウイルスの侵入については、ナファモスタットが1nMで90%以上阻害するのに対して、カモスタットでは同様の阻害に10 nM以上必要であることがわかった。侵入後のウイルス複製に対する阻害能を見ると、ナファモスタットが10 nMで90%以上阻害するのに対して、カモスタットでは、同様の阻害に100 nM以上必要であることがわかった。これらの結果から、生MERSコロナウイルスの感染においても、膜融合と同様にナファモスタットがカモスタットより10倍以上活性が強いことが明らかになった。また、膜融合の実験系での薬物の阻害濃度は、実際の生ウイルスの感染阻害濃度とほぼ同程度あることもわかった。このことは、生ウイルスの実験がBSL3で実施され、かつ実験手技においても高レベルが要求されるのに対して、膜融合系では生ウイルスを使わなくても、生理的に近い条件で阻害剤のスクリーニングができることを示している。すなわち、本融合系の汎用性、利便性はかなり優れたものであり、今後色々な解析に応用可能になると考えている。これらの一連の成果は、山本先生を筆頭著者として2016年8月に発表している[12]。その後、この融合系を用いて、約1万化合物をスクリーニングしたが、ナファモスタット を超える活性を持つ化合物は見つかっていない。この時はMERSの患者は国内におらず、すぐに臨床試験を実施する環境ではなかったので、この段階でコロナウイルスから離れ、別の実験を開始した。
| 図表9 |
| HTSの結果 |
| ここでは融合系の結果のみを提示しているが、たとえ融合が通常に起きても、ルシフェラーゼの酵素活性自体を抑制する化合物も、一見融合を抑制したかのように見える。そのような偽陽性をチェックするために、293FT細胞にSタンパク質は発現させず、DSP1-7とDSP8-11を共発現させ、予め再構成したルシフェラーゼを発現した細胞を作成し、化合物添加により発光が低下しないかを確認する実験をしている。このようなコントロール実験を、筆者らは“コトラ実験”と呼んでいる(DSP1-7とDSP8-11をco-transfection(コトラ)するので)。ナファモスタット は融合系では発光を強力に抑えるが、コトラ実験での発光にはほとんど影響を与えない。 |
 |
| 図表10 |
| 臨床で使われているセリンプロテアーゼ阻害剤とナファモスタットのMERS-CoV Sタンパク質による細胞融合に対する阻害能の比較 |
| Gabe:ガベキセート、Sive:シベレスタット、Riva:リバーロキサバン、Tela:テラプレビル、Ulina:ウリサスタチン、Sime:シメプレビル。ナファモスタットとカモスタットが群を抜いて強力なのがわかる。 |
 |
| 図表11 |
| ナファモスタットとカモスタットのMERS-CoV Sタンパク質による細胞融合に対する阻害能の比較 |
| ターゲット細胞を気道細胞由来のCalu-3に変えると、ナファモスタットもカモスタットも、293FT細胞に比べて10倍程度低い濃度で同程度の阻害能を示す。正確な理由はわからないが、293FT細胞の場合、TMPRSS2とCD26を遺伝子導入により異所性に過剰発現しているためと考えている |
 |
| 図表12 |
| ナファモスタットとカモスタットのMERS-CoVのCalu-3細胞への感染に対する阻害能の比較 |
| この実験では、ウイルス感染前に各薬剤で1時間細胞を処理してからウイルを感染させて6時間後のRNAを回収し、細胞内のウイルスを定量している。6時間後では、増幅したウイルスの再感染は顕著でないと考えられるので、この実験では主にウイルスの侵入に対する薬物の影響を見ている。ここでは示さないが、ウイルス感染後に薬物を加え、その24時間後の培養中のウイルスを定量する実験もしており、その実験においても両薬物は同程度の阻害能を示している。即ち、培養中に繰り返し起こる感染も抑えていると考えられる。 |
 |
SARS-CoV-2のパンデミックが起きて
昨年暮れに武漢でSARS-CoV-2が原因となるCOVID-19の患者が確認されてから、今年に入り感染者は国内でも確認され、その数は世界中で瞬く間に増えていった。SARS-CoVやMERS-CoVと異なり、感染者でも無症状や軽症の感染者が多いことが、このウイルスの感染拡大に拍車をかけていた。感染者の一部は、重症化し肺炎に止まらず、サイトカインストームや血管内皮細胞へのウイルス感染に伴う血栓形成による全身症状、多臓器不全を経て死に至る場合もある。1月10日ごろにSARS-CoV-2の塩基配列が中国のグループから発表され、SARS-CoVやMERS-CoVとのSタンパク質の構造比較から、受容体はSARS-CoVと同じACE2であり、TMPRSS2によりプライムされることが予想された(論文としては[13]に掲載されている)。おそらく世界中の関連研究者がそう予想し走り出したと思う。ただ、中国国内や他国でも一部の研究者は、それ以前に情報を得ているであろうと想像されたので、自分たちもかなり急がないと間に合わない、特に治療薬は既存薬から検討し始めるであろうし、ナファモスタットは筆者らが4年前にMERS-CoVで発表していることもあり、すぐに試されてしまうだろうとかなり焦っていた。予想通り1月31日にbioRxivというpre-print serverに、ドイツのHoffmannらからACE2が受容体で、TMPRSS2がプライミングすると報告された(後にこの論文はCellに掲載された)[14]。
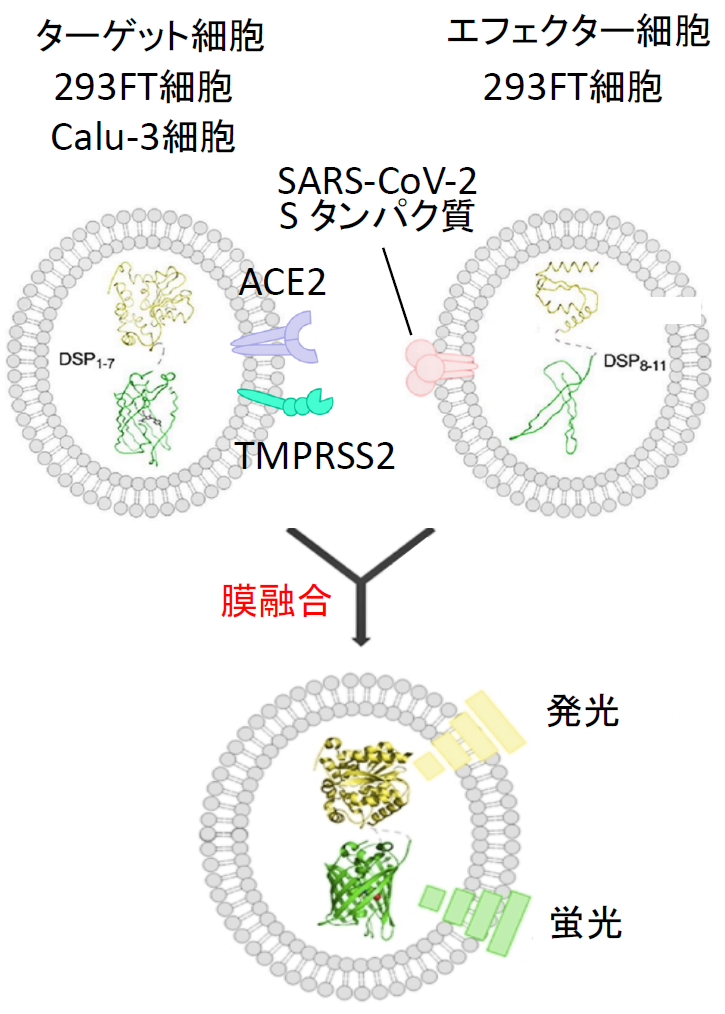
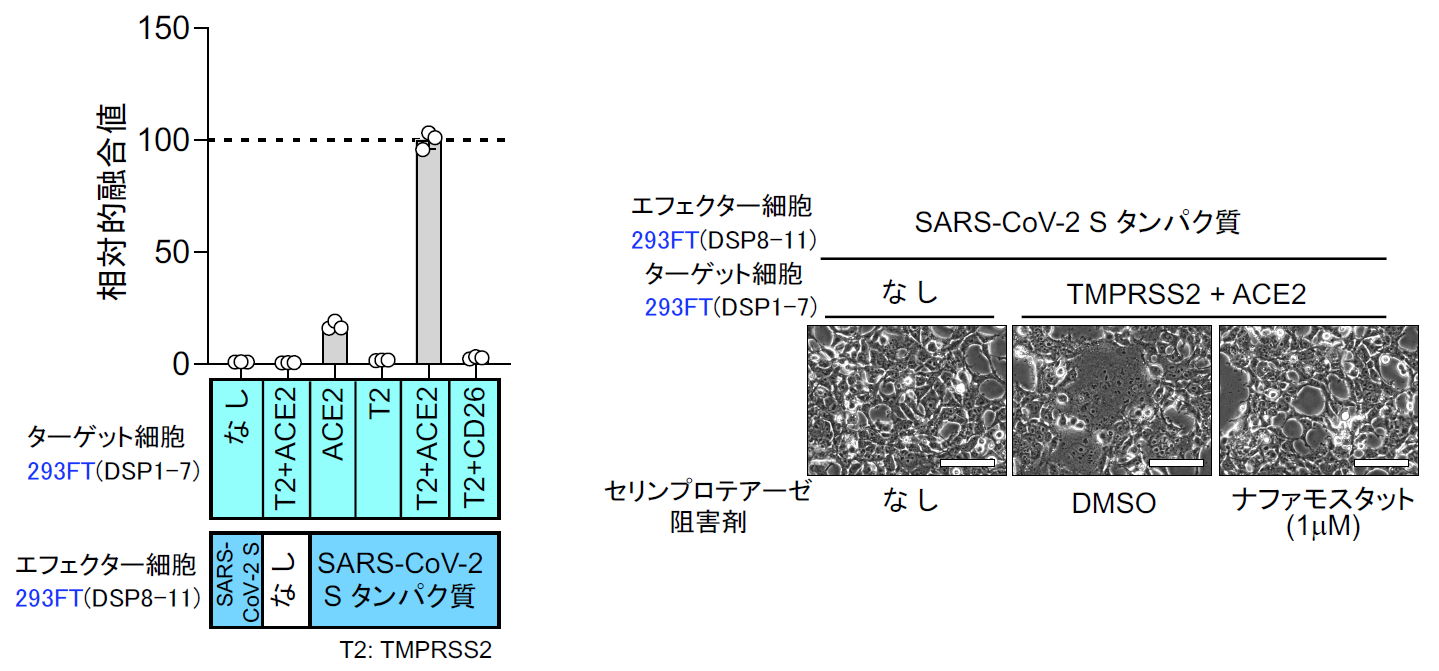
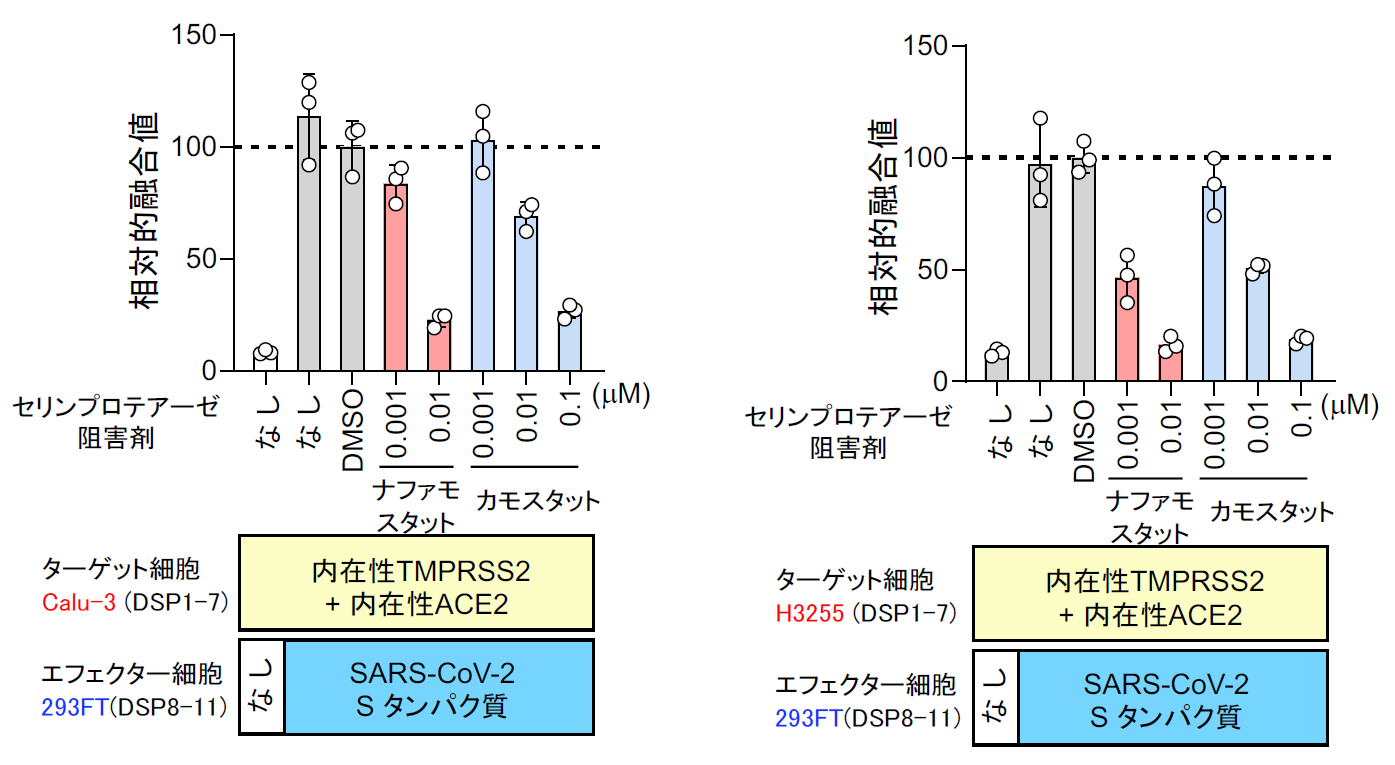
また2月1日には、ナファモスタットのウイルス感染阻害能に関するデータが、中国のWangらによりCell Researchに掲載された[15]。この論文では7種類に既存薬を試していて、レミデシビルとクロロキンは有効であるが、ナファモスタット はEC50=22.5 μM で非常に効果が低いという結果であった。4年前の自分たちのMERS-CoVの結果から、SARS-CoV-2がMERS-CoVと根本的に異なるウイルスでない限り、EC50はnMオーダーと筆者らは予想していたので、何故なのか首を傾げていた。この謎は、あとで解けるのであるが、この時点では、まず自分たちの研究の出発点である膜融合系で、SARS-CoV-2のSタンパク質による膜融合に対するナファモスタット の効果を確認することにした[16]。SARS-CoV-2 Sタンパク質及びDSP8-11を293FT細胞に発現させたエフェクター細胞、およびACE2とTMPRSS2及びDSP1-7を293FTに発現したターゲット細胞を作成した【図表13】。両細胞を混ぜると効率よく融合が起こり、この融合がSタンパク質、受容体ACE2、TMPRSS2に完全に依存する特異的な細胞融合であった【図表14】。
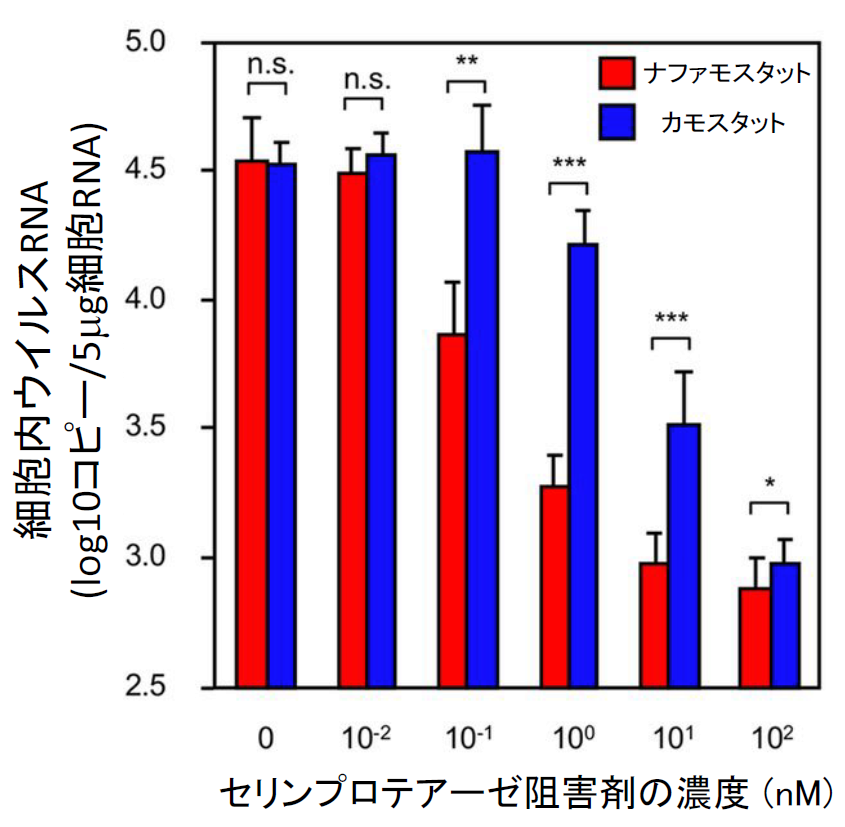
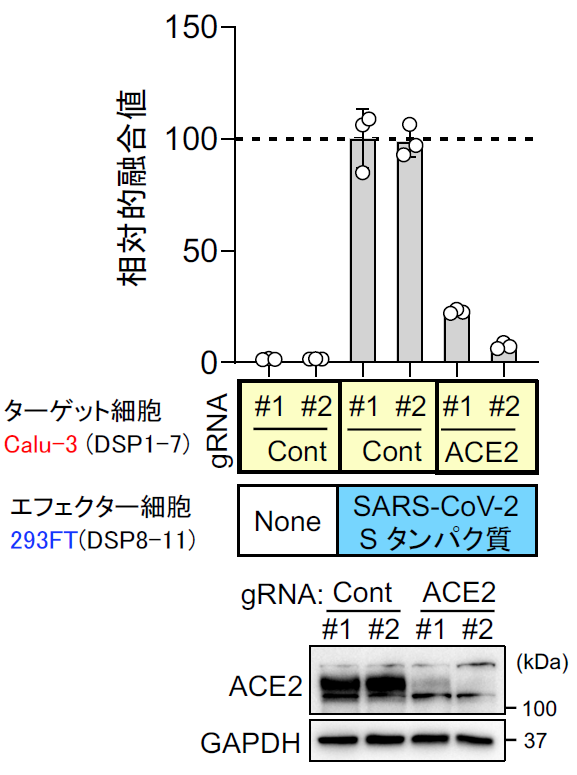
実際に位相差顕微鏡で見ても、ACE2、TMPRSS2依存的に細胞同士の融合が起こりナファモスタット 添加でその融合が抑制されているのがわかる【図表14】。次にMERS-CoVと同様ターゲット細胞を、より生理的条件に近く内在性にACE2とTMPRSS2を発現しているヒト軌道細胞由来のCalu-3細胞を用いたが、やはり効率よく融合が起こった。ACE2を欠損させたCalu-3細胞では融合が起こらないことから、特異的な融合と判断できた【図表15】。次にナファモスタット とカモスタットを加えたところ、MERS-CoVのSタンパク質による融合と同様にナファモスタットは10 nMで、カモスタットは100 nMで80%以上の阻害を示した。この結果は、別のヒト気道上皮由来細胞であるH3255細胞でも再現された【図表16】。ナファモスタットが10 nMで融合を顕著に抑えること、及びナファモスタットはカモスタットに比べて10倍強力であることは、SARS-CoV-2とMERS-CoVの場合と全く同じ結果であった。であれば、実際のウイルス感染もnMオーダーで抑制することが期待される。
| 図表13 |
| SARS-CoV-2 Sタンパク質による細胞融合系 |
| SARS-CoV-2 SタンパクおよびDSP8-11を293FT細胞に発現させてエフェクター細胞を作成し、一方で受容体であるACE2とSタンパク質を活性化するTMPRSS2およびDSP1-7を発現させたターゲット細胞を作成する。両細胞を37℃で混合すると細胞融合が30分以内に起こり始める。 |
 |
| 図表14 |
| SARS-CoV-2 Sタンパク質による細胞融合の特異性 |
| SARS-CoV-2 Sタンパク質による細胞融合系は、Sタンパク質、受容体ACE2、TMPRSS2の全てに依存する特異性を有する(左図)。SARS-CoV-2 Sタンパク質による融合とそのナファモスタットによる抑制を位相差顕微鏡で観察した。 |
 |
| 図表15 |
| ターゲット細胞としてCalu-3細胞を用いた場合のSARS-CoV-2 Sタンパク質による細胞融合におけるACE2の必要性 |
| 内在性にACE2とTMPRSS2を発現するCalu-3細胞における融合の特異性を確認するため、ACE2ノックアウト細胞を2系統作成した。 |
 |
| 図表16 |
| SARS-CoV-2 Sタンパク質による細胞融合に対するナファモスタットとカモスタットの阻害能の比較(気道上皮細胞由来Calu-3 およびH3255細胞) |
| MERS-CoVの場合と同様に、ターゲット細胞を気道細胞由来のCalu-3やH3255細胞に変えるとナファモスタットもカモスタットも293FT細胞に比べて10倍程度低い濃度で、同程度の阻害能を示した。 |
 |
ナファモスタットの抗ウイルス作用は細胞特異的
SARS-CoV-2に感染実験は、医科研の河岡義裕教授との共同研究で、河岡研の木曽真紀先生にご協力いただいた。感染させる細胞として気道細胞由来のCalu-3細胞と、アフリカミドリザルの腎臓由来のVeroE6細胞に異所的に遺伝子導入によりTMPESS2 を発現させたVeroE6/TMPRSS2細胞を用いて、ナファモスタット による感染阻害のEC50を調べた【図表17】。ウイルスを感染させる前に細胞を1時間ナファモスタット で処理して(前処理あり)、その後ウイルス感染30分、さらにその後同濃度のナファモスタット を含む培養液と交換した場合、EC50はCalu-3細胞でおよそ10 nM、VeroE6/TMPRSS2細胞で31.6 μMであった。一方、ナファモスタットをウイルス感染後に添加する(前処理なし)場合、Calu-3細胞で3.16 μM、VeroE6/TMPRSS2細胞では100 μM以上という結果であった。ここでまず重要なことは、Calu-3細胞の場合、予めウイルス感染前にナファモスタット が存在すれば 10 nM程度で感染を抑制できることである。前処理なしで阻害効果が減少するのは、ナファモスタット の作用点がウイルス侵入であるところが大きいと考えられる。実際の患者への投与を想定すると、体内での感染拡大の抑制に効果があると考えられる。
| 図表17 |
| ナファモスタットのSARS-CoV-2のVeroE6/TNPRSS2細胞及びCalu-3細胞への感染に対する阻害能 |
| Calu-3細胞とVeroE6/TMPRSS2細胞のナファモスタット に対する感受性の違いについては、本文に説明した。 |
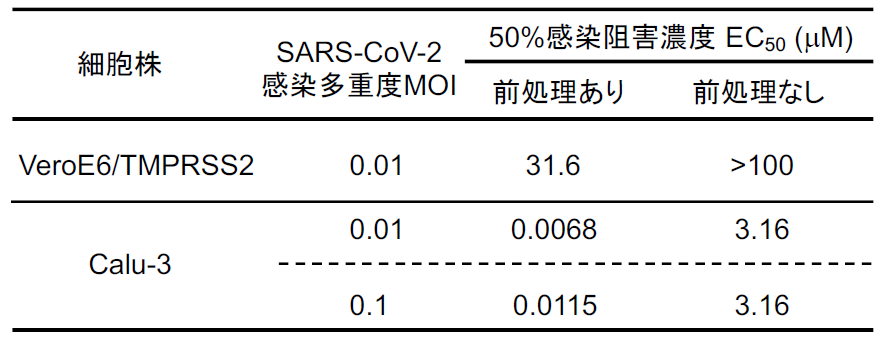 |
さて、VeroE6/TMPRSS2細胞は前処理ありでも、EC50=31.6 μMで阻害効果がCalu-3に比べて著しく弱い。2月初めにWangらがCell Researchに発表したVeroE6細胞用いたナファモスタット のEC50=22.5 μMとほぼ同じ値である[15]。ここで、2月初めにWangらの論文に首を傾げた謎「なぜナファモスタット がSARS-CoV-2の感染をCalu-3細胞では効果的に抑えるのに、VeroE6細胞では効果が低いのか」が以下のように説明できる。そもそもコロナウイルスには、ウイルス外膜と細胞膜との融合を介するTMPRSS2依存的な侵入経路と、ウイルスがエンドサイトーシスされてからウイルス外膜とエンドゾーム膜との融合を介するTMPRSS2非依存的でカテプシンに依存する侵入経路の2種類が存在する[7]。どうも細胞によってこの二つの感染経路を使うバランスが異なるらしい。VeroE6細胞にはTMPRSS2が発現していないこともあり[17]、ウイルスの侵入のほとんどは、TMPRSS2非依存的なエンドゾーム経路を介する。したがってナファモスタット の効果は見えにくい。TMPRSS2を異所的に発現させたVeroE6/TMPRSS2細胞でも、エンドゾーム経路が優勢であることがわかっているので[18]、この細胞でもナファモスタットの効果は見えにくい。一方でCalu-3細胞では、ほとんどがTMPRSS2依存的な細胞膜からの侵入経路を介する[12, 14, 19]ので、ナファモスタットの効果が顕著に現れてくると解釈できる。
ナファモスタットの効果がCalu-3細胞で顕著であるのは、筆者らの他にもドイツや韓国のグループからも報告されている[20, 21]。すなわちナファモスタットの抗ウイルス作用は細胞特異的といえる。ヒト気道由来Calu-3細胞だけでなく、ヒト肺上皮初代培養細胞でもTMPRSS2依存的な細胞膜からの侵入経路が主であることが報告されている[14]。さらに、ヒトでの感染が報告されている鼻腔上皮細胞、肺上皮細胞、腸細上皮胞でACE2とTMPRSS2が共発現している[22, 23]ことは、in vivoでのTMPRSS2依存的侵入経路の重要性を示唆している。またSARS-CoVやMERS-CoVを野生型とTMPRSS2欠損マウスに感染させた実験では、ウイルス感染による体重減少や肺で観察される病理的異常が、TMPRSS2欠損により顕著に減少することが報告されている[24]。これらの報告を総合すると、TMPRSS2依存性侵入経路がin vivoでのSARS-CoV-2の感染や病原性に重要であることが想定され、そしてナファモスタット は、このTMPRSS2依存性侵入経路を感染の主経路としている細胞でのウイルス感染を効果的に抑制すると考えられる。ヒト気道由来Calu-3細胞使った感染実験は非常に重要で、筆者らの2016年のMERS-CoV研究や今回のSARS-CoV-2研究がなければ、ナファモスタット の治療薬としてのポテンシャルを見過ごしてしまう所だったと言える。
ナファモスタットの薬物動態
一方でナファモスタット は既存薬として長い歴史があり、薬物動態はよく調べられている。急性膵炎の場合、ナファモスタットを10 mgあるいは20 mgを90分間かけて1日1〜2 回点滴静注する。この時、血中濃度は点滴開始後90分で30 nMあるいは114 nM達するが、1時間後には9 nM以下になりその後消失する[25]。In vitroの感染阻害EC50が10nM程度であるが、血中濃度が持続しなければ効果は期待できない。一方で、播種性血管内凝固症候群disseminated Intravascular coagulation (DIC) 治療の場合、0.1から0.2 mg/kg体重/時間で2週間程度持続的に点滴静注する。0.2 mgの条件だと血中濃度は26 nM(初期値)から241 nM値を推移し、平均140 nM程度を維持する[25]。
血漿タンパク質との結合率66.64%であること[26]を考慮し、結合状態では効果を発揮しないと仮定しても、約50 nMが有効濃度と考えられる。In vitroのEC50=10 nMであるので、ナファモスタットを抗ウイルス剤としてin vivoでも効果を発揮することが十分期待できると考えた。さらにラットを使った実験ではあるが、ナファモスタットを投与した時、肝臓、腎臓、肺、膵臓の濃度は血中濃度の約45倍、160倍、60倍、6倍となり、かつ長時間の持続することが報告されている[27]。人の場合、同程度の蓄積があるか不明であるが、肺胞の上皮細胞付近での濃度がμMオーダーに到達する可能性があり、ナファモスタットのin vivoでの感染阻害効果はかなり現実的である。
臨床でのナファモスタット:抗ウイルス剤 + 抗凝固剤 + ?
ナファモスタット のCOVID-19に対する臨床試験は、東大病院を中心に5月1日より開始した[28]。世界的には、イタリア、イギリス、インド、韓国でも開始されている。臨床試験においては、抗ウイルス作用はもちろんのこと、他にもナファモスタットの効果として注目したい点がある。
最近の研究からCOVID-19は肺炎の症状から始まるものの、かなり複雑な病態を辿って重症化することがわかっている。特に鍵となる病態としてサイトカインストームと血栓形成が知られている。
ナファモスタット はもともと、トロンビン、活性型凝固因子XIIa, Xa, VIIa, 血漿カリクレイン、プラスミン等のセリンプロテアーゼを阻害することで、線溶系亢進型DICの治療に使われてきた[25, 26]。
したがって、ナファモスタット はCOVID-19における血栓形成を抑制し、重症化を防ぐ可能性がある。この点は臨床試験に際には確認したいところである。
また、マウスの喘息モデルでは、炎症の鍵となる転写因子NF-κBを抑制して、サイトカイン産生を抑制するという報告もある[29]。すなわちサイトカインストームも抑制する可能性はある。こちらの方も臨床試験においては、気になる項目である。抗ウイルス作用を含めて、このようなCOVID-19治療薬としての効果は、大いに期待できると考えているが、あくまで今後の臨床試験のなかで検証される必要がある。
基礎研究のポテンシャル -メリットとデメリット-
ナファモスタットは基礎研究が生んだシーズであり、COVID-19の治療薬の候補となっている。
膜融合定量系の開発という基礎研究から、可能性のあるシーズが生まれたことは、橋渡し研究の一端を担えたと考えている。
もちろん、生ウイルスと細胞との融合は、細胞同士の融合と異なる点は多々あることは想像に難くない。つまり膜融合定量系が生理的な状況を反映していない可能性があるというデメリットがある。だが、一方で生ウイルス研究ではBSL-3環境が必須であるが、膜融合定量系の場合、通常の実験室で何千から何十万という化合物をスクリーニングが比較的短時間で終了するメリットは非常に大きい。
このメリットを最大限生かして、さらに化合物の探索以外にもCOVID-19の診断や治療法開発に利用することを想定して、現在さらなる研究を進めている。筆者らの融合系の譲渡を希望するリクエストは国内外を問わず多い。使い方によっては大きなポテンシャルを持っている。それが基礎研究から生まれたこの膜融合系の強みだと考えている。
最後に
現在、国内外では複数の既存薬がそれぞれの基礎研究から提唱され、臨床試験で評価されようとしている。ナファモスタット以外にも、ファビピラビル、シクレソニド、トシリズマブ、カモスタット、イベルメクチン等が期待されているが、いずれも確固たる基礎研究の成果がその背景にある。
作用点の異なる抗ウイルス薬の併用、重症化を誘導するサイトカインストームや血栓形成を抑制する薬の併用等が予想される標準治療と個人的には考えているが、まだゴールは見えていない。
SARS-CoV−2やCOVID-19に関する研究成果は、基礎、臨床を問わず毎日膨大な数が発表されている。その中には、治療法開発の鍵となるヒントが必ずあるはずで、それを見逃してはならないと思っている。本稿に述べた研究は、多くの共同研究者や協力者によって進んできた。また、ナファモスタット に関する4年前のMERS-CoVの研究と今回のSARS-CoV-2の研究は、いずれも医科研アジア感染症研究拠点 山本瑞生先生が中心になり進めた成果である。
[引用文献]
- Coronavirus disease (COVID-19) outbreak situation. WHO HP https://www.who.int/emergencies/diseases/novel-coronavirus-2019
- 国内の発生状況 厚生労働省HP https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/0000164708_00001.html#kokunaihassei
- 東京大学医科学研究所HPアジア感染症研究拠点 http://www.rcaid.jp
- J. Biol. Chem. 2010 May 7;285(19):14681-8.
- Protein Eng. Des. Sel. 2012 Dec;25(12):813-20.
- 中東呼吸器症候群(MERS)のリスクアセスメント(2019 年 10月 29日現在) 国立感染症研究所HP https://www.niid.go.jp/niid/images/epi/mers/mers-ra-191029.pdf
- Biochimie. 2017 Nov; 142:1-10.
- Proc Natl Acad Sci USA. 2020 May 26;117(21):11727-11734.
- Nature 2013 Aug 8;500(7461):227-31
- Virus Res. 2015 Apr 16; 202:120-34.
- Biochim Biophys Acta 1981 Oct 13;661(2):342-5.
- Antimicrob Agents Chemother 2016 Oct 21;60(11):6532-6539.
- Nature 2020 Mar;579(7798):265-269.
- Cell 2020 Apr 16;181(2):271-280.e8.
- Cell Res. 2020 Mar;30(3):269-271.
- Viruses 2020 Jun 10;12(6): E629.
- Proc Natl Acad Sci USA. 2020 Mar 31;117(13):7001-7003.
- BioRxiv 2020 https://doi.org/10.1101/2020.03.11.987016
- J Virol. 2012 Jun;86(12):6537-45.
- Antimicrob Agents Chemother. 2020 May 21;64(6): e00754-20.
- BioRxiv 2020 https://doi.org/10.1101/2020.05.12.090035
- Cell 2020 May 28;181(5):1016-1035.e19.
- Nat Med. 2020 May;26(5):681-687.
- J Virol. 2019 Mar 5;93(6): e01815-18.
- 医療用医薬品: フサン https://www.kegg.jp/medicus-bin/japic_med?japic_code=00048710 (accessed on 7 June 2020).
- 医薬品インタビューフォーム:フサン https://www.nichiiko.co.jp/medicine/file/31050/interview/31050_interview.pdf
- 基礎と臨床 1984 18(8) 546-562
- 新型コロナウイルス感染症(COVID-19)に対する治療法の早期確立を目指す 東京大学HP https://www.h.u-tokyo.ac.jp/press/__icsFiles/afieldfile/2020/05/08/release_20200508.pdf
- J Allergy Clin Immunol. 2006 Jul;118(1):105-12.